
Perspectives sur les souches du SRAS-CoV-2 | ORF – Bien choisir son serveur d impression
Le SRAS-CoV-2, le virus responsable de la pandémie en cours, évolue à mesure qu'il se propage dans le monde. Cependant, les affirmations concernant une souche plus agressive se propageant à travers les populations humaines ne sont que des conjectures à ce stade. Il est nécessaire de mener des études rigoureuses qui couplent les données cliniques (telles que les caractéristiques des patients et les résultats) avec les changements du virus, ainsi que des études de laboratoire qui testent l'effet des mutations sur la capacité du virus à se répliquer et à se propager. Sans ces preuves, il est spéculatif d'attribuer la propagation et la gravité de la maladie aux mutations. Ce mémoire examine les affirmations selon lesquelles les changements dans le génome du virus SARS-CoV-2 le font se propager plus rapidement ou augmentent sa virulence.
Attribution: Chitra Pattabiraman, Farhat Habib et Krishnapriya Tamma, «Perspectives sur les souches du SRAS-CoV-2», Dossier thématique ORF n ° 365, Mai 2020, Observer Research Foundation.
Sommaire
introduction
La pandémie en cours de la maladie à coronavirus 2019 (COVID-19) est causée par le syndrome respiratoire aigu sévère Coronavirus 2 (SRAS-CoV-2).[1] Il s'agit d'un virus à ARN (acide ribonucléique), avec un génome de longueur 29 903 paires de bases (voir encadré 1). Le génome est organisé en 11 gènes qui codent pour différentes protéines (voir figure 1), telles que la protéine Spike.[2]
Des chercheurs du monde entier ont séquencé le génome du virus en utilisant des échantillons d'individus infectés. Début mai 2020, plus de 17 000 génomes étaient disponibles dans des bases de données publiques (GISAID, NCBI).[3],[4] Ce partage des données en temps opportun a permis aux chercheurs de comparer ces séquences,[5] révélant des différences entre les séquences survenues en raison de mutations (voir l'encadré 1). La plupart des mutations n'entraînent pas de modifications du virus; certains peuvent être délétères et d'autres entraînent des changements positifs. Les mutations doivent être prises en considération lors de la conception de moyens de détecter un virus, de développer des vaccins ou de tester des médicaments potentiels, car certaines mutations peuvent conduire à des faux positifs dans les tests de diagnostic, à une absence de réponse au vaccin et au développement d'une résistance à un médicament. . Les mutations qui modifient la propriété du virus peuvent donner naissance à des variantes biologiquement distinctes appelées «souches» (voir encadré 5). Au moment de la rédaction de ce mémoire, rien ne prouve que les séquences du SRAS-CoV-2 présentent des différences biologiques, ce qui indique qu'une seule souche de ce virus est actuellement en circulation. [6]
Figure 1: Organisation du génome du SRAS-CoV-2

Remarque: Les gènes sont représentés en jaune, les peptides résultants sont affichés en vert, les régions non traduites sont affichées en rouge et violet. Une première séquence de Wuhan, en Chine (NC_045512) a été utilisée pour réaliser ce schéma à l'aide de Geneious (Ver. 2020.0).
Les «types» S et L du SRAS-CoV-2
En février 2020, Tang et al. a publié une analyse de 103 génomes du SRAS-CoV-2.[7] Les génomes ont été regroupés en deux groupes, sur la base de deux mutations. L'un d'eux était un changement d'acides aminés (de la sérine à la leucine) en position 84 (voir l'encadré 3) dans la région ORF8, provoquant la production d'une protéine légèrement différente. Les deux groupes de génomes étaient étiquetés de types S (sérine) et L (leucine). Le type L était plus répandu parmi les virus étudiés. Sur cette base, les chercheurs l'ont qualifié de «type agressif». Cependant, cette classification a été remise en question par la communauté scientifique,[8],[9] et Tang et al. a concédé que sans études cliniques et / ou de laboratoire suffisantes pour confirmer l'hypothèse, «fréquence plus élevée» était un terme plus approprié que «agressif». Malheureusement, les dégâts ont déjà été causés car l'article original a été repris par les médias internationaux.
Un SARS-CoV-2 «mutant»
Vers la fin avril 2020, Korber et al. analysé 4 535 génomes SARS-CoV-2 accessibles au public (GISAID) et présenté leurs résultats sur le serveur de pré-impression, bioRxiv.[10] Ils se sont concentrés sur la partie du génome qui code pour la protéine Spike (voir figure 1), qui interagit avec son récepteur sur les cellules humaines, permettant au virus d'entrer dans la cellule (voir encadré 4).[11] Les chercheurs ont découvert qu'à la position 614 de la protéine, il y avait un changement de l'acide aspartique à la glycine (D614G), par rapport à la souche de référence, résultant en une variante de la protéine Spike. De plus, la fréquence de cette mutation augmentait à travers le monde. Étant donné que la mutation est sur la protéine qui facilite l'entrée du virus dans la cellule hôte, les chercheurs ont postulé que ce changement pourrait influencer la capacité de la protéine à se lier au récepteur ACE2 et que la mutation pourrait avoir un impact sur la façon dont le virus est vu et reconnu par le système immunitaire. Bien qu'il s'agisse d'hypothèses valables, elles ne sont actuellement pas testées. Enfin, leurs analyses indiquent que lorsque des virus contenant cette mutation entrent dans une population, ils deviennent le virus dominant dans cette région.
Le consensus actuel sur les souches du SRAS-CoV-2
Une «souche» pour un virus (voir encadré 5) doit avoir, à tout le moins, des propriétés biologiques distinctes, par ex. différents taux de croissance et stabilité dans différents environnements. Ainsi, les isolats viraux du SRAS-CoV-2 ne sont pas admissibles, car ils n'ont aucune différence biologique documentée.[12]
Cependant, l'analyse des données de séquençage mondiales a suggéré que, sur la base de la différence de séquence, les génomes du SRAS-CoV-2 peuvent être classés en au moins 10 groupes distincts. Un arbre phylogénétique illustre les relations entre les différentes séquences (voir Fig. 2). Un clade est un groupe de séquences hautement apparentées qui partagent un ancêtre commun, c'est-à-dire qu'elles font partie d'une branche. Dans la phylogénie du SRAS-CoV-2, de nombreux isolats originaires de Chine font partie du clade B. Les isolats du monde entier appartiennent au clade A (en utilisant la même nomenclature que celle utilisée par nextstrain.org). La phylogénie est mise à jour à mesure que de nouvelles séquences arrivent, et l'analyse la plus récente (début mai 2020) des données (5234 génomes complets) par 386 groupes à travers le monde montre que l'A2a est le clade le plus séquencé (voir figure 2). L'une des mutations définissant le clade A2a est la même que celle décrite par Korber et al., C'est-à-dire D614G sur la protéine Spike.
Majumder et al. sont parvenus à une conclusion similaire sur la prévalence mondiale de A2a (résultats non publiés).[13] Cependant, le Dr Majumder a déclaré dans une interview que les données disponibles en provenance d'Asie ne montrent pas une nette domination du clade A2a.[14]
Figure 2: Phylogénie de 5 234 génomes complets
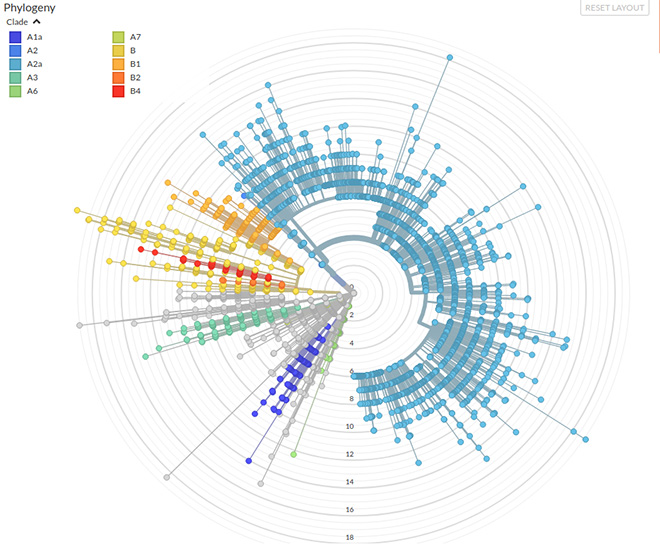
Transmission A2a Clade
Quatre facteurs principaux limitent la capacité des chercheurs à déterminer si le clade A2a transmet mieux que les autres clades. Ces facteurs doivent être pris en compte lors de l'analyse des taux de transmission de différents clades de SARS-CoV-2 dans le contexte mondial.
- Les paramètres contribuant à la propagation varient avec le temps et sont influencés par des facteurs culturels qui influent sur le respect des mesures d'atténuation. Il est donc difficile d'estimer le taux de croissance des infections. La propagation d'un virus dans une région est affectée par le calendrier et le type de mesures d'atténuation mises en place.[16] Les pays qui ont retardé la mise en œuvre de telles mesures pour le SRAS CoV-2 verront une fréquence plus élevée de la souche du virus qui a d'abord infecté la population. Ce phénomène est connu sous le nom d '«effet fondateur» et peut déterminer quel clade devient dominant. Dans une telle situation, la nature réelle de la mutation qui a donné naissance à ce clade est sans importance.
- Les estimations des paramètres des modèles de prédiction ne sont pas fiables aux premiers stades d'une épidémie, en raison de la petite taille des échantillons sur lesquels ils sont fondés. Cela se reflète dans la variation entre les prévisions de différents modèles concernant le nombre total d'infections. Par exemple, selon les prévisions des États-Unis et du CDC publiées au cours de la deuxième semaine d'avril, le meilleur scénario avec une réduction de 20% des contacts était d'environ 200 000 décès aux États-Unis la première semaine de mai.[17] Plus tôt, un rapport de l'Imperial College (Londres), en mars, avait produit une prévision basée sur la modélisation qui prévoyait environ 2,2 millions de décès aux États-Unis d'ici août 2020, avec un pic en juin, si les mesures restaient inchangées.[18] Estimations du taux de reproduction de base, R0, variait également largement mais variait généralement entre 2,2 et 3,9.[19]
- Des facteurs épidémiologiques tels que l'âge, le sexe, l'accès aux soins cliniques, les conditions comorbides, la transmission asymptomatique et la disponibilité des tests sont susceptibles de jouer un rôle important dans la transmission. Cela empêchera les chercheurs de conclure si un clade est dominant en raison des changements de séquence seuls.
- Les fréquences relatives des différents clades déduites des données de séquençage dépendront de la capacité de séquencer et de publier les données dans des bases de données publiques. Par exemple, sur les quelque 17 000 séquences de GISAID au cours de la première semaine de mai, près de 15 000 proviennent des États-Unis, du Royaume-Uni et de l'Australie. Ce type de biais dans l'échantillonnage peut montrer une domination apparente mais inexacte de certains clades.
L'implication d'un virus mutant
Les mutations peuvent affecter l'efficacité des méthodes de diagnostic basées sur la séquence et imposer des restrictions sur la conception du vaccin.[20] L'outil de diagnostic le plus couramment utilisé cible des régions spécifiques du génome, et si une mutation se produit dans cette région, le test peut conduire à un résultat faussement négatif. En outre, les vaccins basés sur la séquence peuvent devoir incorporer la mutation D164G et tenir compte de ses effets. Partout dans le monde, les efforts de séquençage marquent des régions en mutation pour guider le diagnostic et la conception des vaccins.
Les mutations font partie du cycle de vie naturel de tout virus, en particulier les virus à ARN. Les nombreuses mutations observées dans les séquences SARS-CoV-2 sont donc attendues.[21] Cependant, il n'est pas clair si ces mutations sont significatives, c'est-à-dire si elles modifient les propriétés biologiques du virus. Des études visant à évaluer cela doivent encore être menées. S'il est plausible que certaines mutations permettent au virus de se propager plus efficacement, il est prématuré de conclure que ces mutations sont à l'origine de la propagation mondiale actuelle du virus.
Conclusion
Des analyses de séquences de génomes du monde entier ont révélé la présence de différents clades SARS-CoV-2. Cependant, aucune preuve expérimentale ne suggère une différence d'agressivité entre ces derniers. De plus, les effets des mutations observées dans le SRAS-CoV-2 sur les propriétés du virus doivent encore être évalués dans des études cliniques ou expérimentales. Étant donné que plusieurs facteurs influencent la propagation virale, la simple prévalence d'un clade viral ne peut pas être utilisée comme indicateur indirect de traits biologiques tels qu'une transmission accrue et la gravité de la maladie. Certains pays sont plus touchés que d'autres en raison d'un mélange complexe de la biologie du virus et du comportement de la population infectée et sensible. Une liste exhaustive des principaux facteurs ne peut émerger qu'avec le temps, à mesure que davantage de données deviennent disponibles.
Avertissement: Certains des travaux de recherche cités ici n'ont pas encore été examinés par d'autres scientifiques de manière formelle (examen par les pairs). Les informations présentées dans cet article sont à jour au 18 mai 2020.
Encadré 1: Le génome du SRAS-CoV-2Le SRAS-CoV-2 est un virus à ARN, et son génome est constitué d'un arrangement unique de quatre ribonucléotides – Adénine (A), Uracile (U), Guanine (G), Cytosine (C) – d'environ 30 000 bases de long. Le génome peut être considéré comme un long paragraphe. Lorsqu'un virus infecte une cellule, il se copie. Cela nécessite de copier le paragraphe, dans lequel des erreurs (c'est-à-dire des mutations) peuvent se produire. Plus le nombre de copies est élevé, plus les chances d’accumuler des «mutations» sont élevées. Certains d'entre eux peuvent changer le sens, d'autres peuvent rendre le paragraphe vide de sens, tandis que de nombreuses erreurs (telles que les fautes de frappe) peuvent ne pas affecter la signification du tout. |
Encadré 2: Les virus évoluent-ils de manière à mieux transmettre ou à provoquer des maladies plus graves?Virus de la grippe aviaire – Des études ont montré qu'un petit nombre de mutations dans les virus de la grippe aviaire peuvent les faire se transmettre aux mammifères.[22]De plus, un petit nombre de mutations devraient augmenter les chances de propagation aux populations humaines et de transmission entre humains.[23] Il est supposé que quelque chose de similaire s'est produit chez un ancêtre du SRAS-CoV-2, lui permettant de sauter chez l'homme en 2019. SRAS – Lorsque le virus du syndrome respiratoire aigu sévère (SRAS) est apparu en 2003, une délétion de 29 nucléotides a été observée. Il a été initialement supposé avoir un effet positif; cependant, des études ultérieures ont démontré un effet délétère de cette mutation sur le virus.[24] Virus Zika – Il existe deux lignées (africaine et asiatique) du virus Zika transmis par les moustiques, en fonction des différences dans leurs séquences génétiques. Cependant, les lignées africaines ne sont pas connues pour provoquer des malformations congénitales, tandis que les lignées asiatiques ont provoqué des épidémies de microcéphalie dans de nombreuses régions du monde. Il existe des preuves suggérant que la lignée asiatique a acquis une seule mutation, la rendant plus pathogène que les virus Zika plus anciens de la même lignée.[25] Cette souche était responsable de l'épidémie dévastatrice au Brésil en 2015. Virus Ebola – Une mutation du virus Ebola a été signalée en 2015 et aurait provoqué une souche plus agressive.[26] Cependant, bien que les études initiales aient fourni des preuves solides de ces différences, celles-ci n'ont pas été reproduites dans des modèles animaux. Ainsi, il s'est avéré difficile d'interpréter l'effet de cette mutation.[27] |
Encadré 3: Les éléments constitutifs du corpsLes protéines sont les molécules fonctionnelles clés de la biologie: la plupart des enzymes et de nombreuses hormones et toxines sont des protéines. Ce sont des chaînes d'unités plus petites appelées acides aminés. Il y a 20 acides aminés, et chacun a des propriétés chimiques spécifiques – ce qu'il peut se lier, s'il peut se dissoudre dans l'eau, etc. Ainsi, la structure et la fonction d'une protéine sont déterminées par la séquence spécifique et l'identité des acides aminés qui constituent il. Les acides aminés sont codés par les séquences nucléotidiques du matériel génétique. Les mutations non synonymes entraînent une modification de l'acide aminé, contrairement aux mutations synonymes. Les changements dans les acides aminés dans la séquence protéique peuvent changer la structure et la fonction de la protéine. |
Encadré 5: Qu'est-ce qu'une «souche» d'un virus?Une «souche» peut être définie comme une variante d'une espèce virale qui est distinctement reconnaissable et possède certaines caractéristiques phénotypiques uniques, qui sont stables dans des conditions naturelles, par exemple propriétés antigéniques, manifestation de la maladie ou gamme d'hôtes.[30] La condition de «différence génétique stable» exige que les différences de souches soient stables d'une génération à l'autre, probablement en raison de la sélection naturelle. Selon cette définition, si deux virus ne présentent que des différences génotypiques sans différences phénotypiques, ils ne seront pas qualifiés de souches différentes. En revanche, si elles présentent des différences phénotypiques même avec peu de mutations, elles seront qualifiées de souches différentes. Ainsi, l'information génomique seule, bien que très importante, est insuffisante pour déterminer la classification du virus, reconstruire l'évolution ou même comprendre la pathogénicité. |
Encadré 7: Arbre phylogénétiqueUn arbre phylogénétique est une représentation schématique de la relation entre différents taxons. En règle générale, les données génétiques (séquences ou données génomiques) sont utilisées pour reconstruire les arbres phylogénétiques. La forme générale de l'arbre est appelée sa «topologie» et les pointes correspondent aux «taxons» dont la relation est examinée. Celles-ci correspondent souvent à différentes espèces, mais peuvent également correspondre à des souches virales, à des individus d'une espèce, à des genres ou à d'autres niveaux d'organisation plus élevés. Les pointes sont reliées les unes aux autres par des branches. Deux pointes adjacentes sont connectées par un «nœud», qui correspond à l'ancêtre commun commun récent. Les taxons (ou astuces) qui partagent un ancêtre commun sont considérés comme des «sœurs». Si tous les taxons partagent un ancêtre commun et que tous les descendants sont inclus dans le groupe, on l'appelle un «groupe monophylétique». Un composant important d'un arbre phylogénétique est un «clade», qui est un groupe monophylétique de taxons qui comprend l'ancêtre commun le plus récent de tous les membres (c'est-à-dire tous les descendants). Un clade peut être défini à n'importe quel niveau dans un arbre phylogénétique. |
(Les auteurs remercient le Prof. Nagasuma Chandra pour les discussions initiales et la lecture des suggestions de matériel pour ce mémoire. Nous tenons également à remercier les chercheurs du monde entier qui ont déposé des séquences dans GISAID et l'équipe nextstrain.org pour avoir partagé leur analyse.)
à propos des auteurs
Les auteurs sont membres d'un cercle d'étude multidisciplinaire COVID-19 basé en Inde. Chitra Pattabiraman est boursière en début de carrière, India Alliance (DBT-Wellcome Trust), au département de neurovirologie, NIMHANS, Bangalore; Farhat Habib est directeur de la science des données chez TruFactor (une société du groupe InMobi), Bangalore; et Krishnapriya Tamma est professeur adjoint, École des arts et des sciences, Université Azim Premji, Bangalore.
Notes de fin
[1] Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, et al. «Un nouveau coronavirus de patients atteints de pneumonie en Chine, 2019», N Engl J Med (2020). https://doi.org/10.1056/NEJMoa2001017.
[2] Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, et al. «Caractérisation génomique et épidémiologie du nouveau coronavirus 2019: implications pour les origines du virus et la liaison aux récepteurs», Lancette (2020). https://doi.org/10.1016/S0140-6736(20)30251-8.
[3] GISAID. URL: https://www.gisaid.org/
[4] NCBI. URL: https://www.ncbi.nlm.nih.gov/
[5] nextstrain.org. URL: https://nextstrain.org/ncov
[6] "Il existe une et une seule souche de SARS-CoV-2", virology.ws. (2020) URL: https://www.virology.ws/2020/05/07/there-is-one-and-only-one-strain-of-sars-cov-2/
[7] Tang X, Wu C, Li X, Song Y, Yao X, Wu X, et al. «Sur l'origine et l'évolution continue du SRAS-CoV-2», Natl Sci Rev (2020). https://doi.org/ 10.1093 / nsr / nwaa036.
[8] MacLean OA, Orton RJ, Singer JB, Robertson DL. «Réponse à« Sur l'origine et l'évolution continue du SRAS-CoV-2 »(2020). URL: http://virological.org/t/response-to-on-the-origin-and-continuing-evolution-of-sars-cov-2/418/11.
[9] MacLean OA, Orton RJ, Singer JB, Robertson DL. «Aucune preuve de types distincts dans l'évolution du SRAS-CoV-2», Virus Evol (2020);6:. https://doi.org/10.1093/ve/veaa034
[10] Korber B, Fischer W, Gnanakaran SG, Yoon H, Theiler J, Abfalterer W, et al. «Le pipeline de mutation de Spike révèle l'émergence d'une forme plus transmissible de SRAS-CoV-2», BioRxiv (2020).
[11] Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. "L'entrée des cellules du SRAS-CoV-2 dépend de l'ACE2 et du TMPRSS2 et est bloquée par un inhibiteur de protéase cliniquement prouvé," Cellule (2020).
[12] «Il existe une et une seule souche de SARS-CoV-2», virology.ws. (2020).
[13] «Écoutez: la science et les mystères de l'évolution du nouveau coronavirus», 9 mai 2020.
[14] «Ep. 136: The SARS-COV-2 Virus: Mutations & Evolution », 8 mai 2020.
[15] nextstrain.org.
[16] Bruinen de Bruin Y, Lequarre A-S, McCourt J, Clevestig P, Pigazzani F, Zare Jeddi M, et al. «Impacts initiaux des mesures mondiales d'atténuation des risques prises lors de la lutte contre la pandémie de COVID-19», Saf Sci (2020);128: 104773.
[17] Prévisions CDC (2020).
[18] Ferguson N, Laydon D, Nedjati Gilani G, Imai N, Ainslie K, Baguelin M, et al. Impact des interventions non pharmaceutiques (NPI) pour réduire la mortalité par COVID19 et la demande de soins de santé. Dakota du Nord.
[19] Lv M, Luo X, Estill J, Liu Y, Ren M, Wang J, et al. «Maladie à coronavirus (COVID-19): examen de la portée» Eurosurveillance (2020);25: 2000125.
[20] Phelan J, Deelder W, Ward D, Campino S, Hibberd ML, Clark TG. "Contrôle de l'épidémie de SRAS-CoV-2, informations provenant de séquences de génomes entiers à grande échelle générées à travers le monde", BioRxiv (2020): 2020.04.28.066977.
[21] Sidney M. Bell, Emma Hodcroft, Nicola Müller, Cassia Wagner, James Hadfield, Richard Neher TB. "Analyse génomique de COVID-19. Rapport de situation 2020-05-15 ”(2020)
[22] Herfst S, Schrauwen EJA, Linster M, Chutinimitkul S, Wit E de, Munster VJ, et al. «Transmission aéroportée du virus de la grippe A / H5N1 entre les furets», Science (2012);336: 1534–41.
[23] «La reconstruction du virus de la grippe aviaire, semblable à 1918, suscite des inquiétudes au sujet des expériences de gain de fonction»,(2014).
[24] Muth D, Corman VM, Roth H, Binger T, Dijkman R, Gottula LT, et al. "Atténuation de la réplication par une délétion de 29 nucléotides dans le SRAS-coronavirus acquis au cours des premiers stades de la transmission interhumaine", Sci Rep (2018);8: 15177.
[25] Yuan L, Huang X-Y, Liu Z-Y, Zhang F, Zhu X-L, Yu J-Y, et al. "Une seule mutation de la protéine prM du virus Zika contribue à la microcéphalie fœtale", Science (2017);358: 933–6.
[26] Diehl WE, Lin AE, Grubaugh ND, Carvalho LM, Kim K, Kyawe PP, et al. «La glycoprotéine du virus Ebola avec une infectiosité accrue a dominé l'épidémie 2013-2016», Cellule (2016);167: 1088-1098.e6.
[27] Marzi A, Chadinah S, Haddock E, Feldmann F, Arndt N, Martellaro C, et al. «Les mutations récemment identifiées dans le génome du virus Ebola-Makona ne modifient pas la pathogénicité des modèles animaux», Cell Rep (2018);23: 1806–16.
[28] Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. "L'entrée des cellules du SRAS-CoV-2 dépend de l'ACE2 et du TMPRSS2 et est bloquée par un inhibiteur de protéase cliniquement prouvé", Cellule (2020).
[29] Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. "L'origine proximale du SRAS-CoV-2", Nat Med (2020).
[30] Kuhn JH, Bao Y, Bavari S, Becker S, Bradfute S, Brister JR, et al. "Nomenclature des virus au-dessous du niveau des espèces: une nomenclature normalisée pour les variantes naturelles des virus affectés à la famille des Filoviridae", Arch Virol (2013);158: 301.
[31] Louis du Plessis. Signal temporel de nCoV-2019 basé sur 30 génomes, (2020).
[32] Rambaut A. Analyse phylodynamique | 176 génomes | 6 mars 2020 », (2020).
[33] MacLean OA, Orton RJ, Singer JB, Robertson DL. "Aucune preuve de types distincts dans l'évolution du SRAS-CoV-2", Virus Evol (2020);6:.






Commentaires
Laisser un commentaire